class: top, right, title-slide # Integrative statistical analysis of -omics and GWAS data ## Public defense ### Sina Rüeger ### 21 September 2018 --- <!------- emoji 😮----------> <!------- icons <i class="fab fa-r-project " style="color:#88398a;"></i>----------> <!------- icons <i class="ion ion-heart "></i> ----------> --- class: inverse, left, middle # Outline - Introduction - Results - Discussion --- class: inverse, left, middle # Outline - Introduction (= explaining the title) - Results (= what I did during the PhD) - Discussion (= did it work? & outlook) <!---------------- INTRODUCTION ---------------> --- class: inverse, center, middle # Introduction --- class: center, middle ## The goals to keep in mind: *Cornerstones of Medicine* <img src="img/cornerstones.png" width="400"> <!---- Data analysis --------> --- class: center, middle ## 1. What is "data analysis" and "statistics"? --- class: left, middle ### Summarise data #### Example: Distribution of human height in 100 individuals 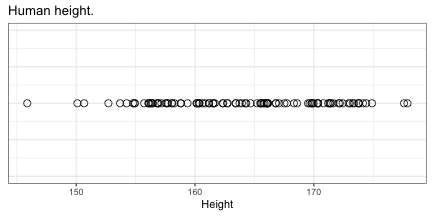<!-- --> --- class: left, middle ### Summarise data #### Example: Distribution of human height in 100 individuals 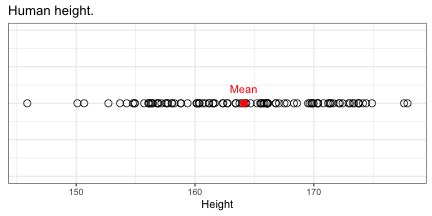<!-- --> - Mean: 164 - Standard deviation = 6.8 --- class: left, middle ### Identify relationships between A and B #### Example: Relationship between age, sex and LDL (low-density lipoprotein) .small[From: Freedman et al. (2004)] <img src="img/age_LDL.png" width="200"> --- class: left, middle ## Central principles of data analysis - Data represents oftentimes an **approximation** for something we cannot measure <i class="fas fa-arrow-right "></i> *Use of BMI as a proxy for obesity*. - The more data records we have, the **more precise** the estimation (of interest) <i class="fas fa-arrow-right "></i> *Relationship between sample size and the standard error*. --- class: left, middle ## Keep in mind <i class="fas fa-hand-pointer "></i> - Statistical methods: **summarise data** & quantify relationships. - Increasing **sample size** helps to estimate precisely. <!---- Biology --------> --- class: center, middle ## 2. Bits of biology or: what is *-omics*? --- class: left, middle ## DNA .pull-left[ - is the blueprint of (human) life - consists of nucleotides A, C, G and T ] .pull-right[ <img src="img/genes-DNA.png" width="800"> .small[Source: https://www.thoughtco.com/genetics-basics-373285] ] --- class: left, middle ## Biological cascade <img src="img/cascade.png" width="800"> --- class: left, middle ## SNP (Single Nucleotide Polymorphism) <img src="img/snps-001.png" width="600"> --- class: left, middle ## SNP (Single Nucleotide Polymorphism) <img src="img/snps-002.png" width="600"> --- class: left, middle .pull-left[ <blockquote class="twitter-tweet" data-lang="en"><p lang="en" dir="ltr">Two of my favourite things put together! <a href="https://t.co/1jE0hWr0Yt">pic.twitter.com/1jE0hWr0Yt</a></p>— Mike Inouye (@minouye271) <a href="https://twitter.com/minouye271/status/993073495205527552?ref_src=twsrc%5Etfw">May 6, 2018</a></blockquote> <script async src="https://platform.twitter.com/widgets.js" charset="utf-8"></script> ] .pull-right[ ## Linkage Disequilibrium (LD) Parts of the **genome tend to be inherited together**, which creates sets of correlated SNPs in close proximity (‘in linkage disequilibrium (LD)’). ] --- class: left, middle ## How DNA is measured <img src="img/genome.png" width="700"> --- class: left, middle ## Keep in mind <i class="fas fa-hand-pointer "></i> - We are only interested in the varying part of the genome <i class="fas fa-arrow-right "></i> **SNPs** - Parts of the genome are correlated <i class="fas fa-arrow-right "></i> **LD** <!---- GWAS --------> <!--- combine DA and biology --------> --- class: center, middle ## 3. GWAS (genome-wide association study) --- class: left, middle ## Relationship between a genotype and trait/disease 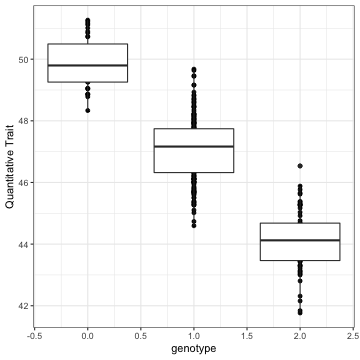<!-- --> --- class: left, middle ## Relationship between a genotype and trait/disease 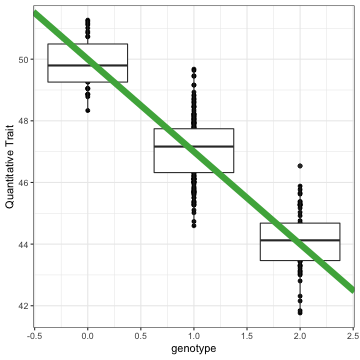<!-- --> --- class: left, middle ## Relationship between a genotype and trait/disease <img src="img/gwas-a.png" width="600"> --- class: left, middle ## Relationship between MANY genotypes and trait/disease <img src="img/gwas-b.png" width="600"> --- class: left, middle ## GWAS summary statistics <img src="img/gwas-ss.png" width="400"> --- class: left, middle ## Large sample sizes needed .pull-left[ <img src="img/maf_effectsize.png" width="400"> ] .pull-right[ - Solution \#1: Exploit genetic correlation and use chips (cheap) instead of sequencing (expensive) - Solution \#2: Form a consortium ] --- class: left, middle ## Example: GWAS in MDD - [Wray et al. (2018)](https://doi.org/10.1038/s41588-018-0090-3) - 44 novel loci associated with major depression disorder (MDD) - a meta-analysis of GWAS summary statistics from 135'458 cases and 344'901 controls from six cohorts. Resulting summary statistic was used: - to identify the somatic tissues involved in MDD - to estimate heritability explained by the new findings - to identify biological pathways relevant to MDD - to investigate the relationships with genetically correlated traits --- class: left, middle ## Keep in mind <i class="fas fa-hand-pointer "></i> - Large sample sizes needed: **consortium**! - Use of GWAS summary statistics with **follow-up methods**. <!---- Missing data --------> --- class: center, middle ## 4. Missing data --- class: left, middle ## How to infer data that is missing? <img src="img/map-arosa.png" width="650"> --- class: left, middle ## How to infer data that is missing? <img src="img/map-arosa-plain.png" width="650"> --- class: left, middle ## Basic principle <img src="img/genome_imputation.png" width="600"> <!---- SSIMP --------> --- class: center, middle ## 5. Summary statistic imputation (SSIMP) --- class: left, middle ## GWAS <img src="img/ssimp-a.png" width="700"> --- class: left, middle ## Untyped variant <img src="img/ssimp-b.png" width="700"> --- class: left, middle ## Genotype imputation <img src="img/ssimp-c.png" width="700"> --- class: left, middle ## Genotype imputation <img src="img/ssimp-d.png" width="700"> --- class: left, middle ## Summary statistic imputation <img src="img/ssimp-e.png" width="700"> --- class: left, middle ## Main equation <img src="img/ssimp-equation.png" width="700"> <!---------------- RESULTS ---------------> --- class: inverse, center, middle # Results --- class: left, middle # Output 1. <i class="fas fa-arrow-right "></i> Evaluation and application of summary statistic imputation <i class="fas fa-arrow-left "></i> - <small>*Evaluation and application of summary statistic imputation to discover new height- associated loci* (Rüeger, McDaid, and Kutalik 2018, PLOS Genetics)</small> 1. Improving summary statistic imputation for mixed populations - <small>*Improved imputation of summary statistics for mixed populations* (Rüeger, McDaid, and Kutalik 2017, BioRxiv)</small> 1. Applications of summary statistic imputation - <small>*Rare and low-frequency coding variants alter human adult height* (Marouli et al. 2017, Nature)</small> - <small>*Bayesian association scan reveals loci associated with human lifespan and linked biomarkers* (McDaid et al. 2017, Nature Communications)</small> 1. Contributions to research in infectious diseases - <small>*Impact of common risk factors of fibrosis progression in chronic hepatitis C* (Rüeger et al. 2015, Gut)</small> - ... 1. Minor contributions to other publications --- class: left, middle <img src="img/rueeger-etal-2018-fp.png" width="800"> --- class: left, middle # Goals 1. **Compare** summary statistic imputation to genotype imputation. 1. **Test** the utility of summary statistic imputation on a real case on human height data. --- class: left, middle # 1. Compare SSIMP to genotype imputation <img src="img/results-ukbb-schema.png" width="600"> --- class: left, middle <img src="img/results-ukbb-associated.png" width="800"> --- class: left, middle <img src="img/results-ukbb-null.png" width="800"> --- class: left, middle # 2. Test SSIMP on height GWAS <img src="img/results-exome-schema.png" width="600"> --- class: left, middle ## Locuszoom plot of results <img src="img/results-exome-locuszoom.png" width="500"> <!---------------- DISCUSSION ---------------> --- class: inverse, center, middle # Discussion --- class: top, middle ## Outlook for SSIMP To impute summary statistics reliably the following is needed ☝️ ### <i class="fas fa-globe " style="color:#E41A1C;"></i> <i class="fas fa-globe " style="color:#377EB8;"></i> <i class="fas fa-globe " style="color:#4DAF4A;"></i> <i class="fas fa-globe " style="color:#984EA3;"></i> <i class="fas fa-globe " style="color:#FF7F00;"></i> Large and diverse reference panels - to improve of LD matrix estimation - to impute low-frequency variants - to impute cosmopolitan/multi-ethnic GWASs -- ### <i class="fas fa-wrench " style="color:gray;"></i> Software & tooling - easy to use and maintained -- ### <i class="ion ion-ios-cloud-download "></i> Publicly available GWAS data - the foundation of SSIMP --- class: left, middle ## Conclusion <i class="fab fa-pagelines " style="color:green;"></i> With my work on summary statistic imputation I have helped to **link incomplete GWAS** summary statistics with **follow-up methods** (such as multi-trait analyses). <!--------- - **Improved SSIMP**: .small[improved imputation quality and optimised assembly of the LD matrix.] - Quantified **performance & utility of SSIMP**: .small[compared to genotype imputation, identified groups of genetic variants that are hard to impute, and demonstrated in a case study the utility of summary statistic imputation.] -----> --- class: inverse, center, middle .big[<font face="Yanone Kaffeesatz"> Thank you! </font>] <!------<i class="fas fa-smile "></i> ----------> Slides: [https://sinarueeger.github.io/publicdefense/slides#1](https://sinarueeger.github.io/publicdefense/slides#1) Source code: [https://github.com/sinarueeger/publicdefense/](https://github.com/sinarueeger/publicdefense/)