R-packages used:
library(httr)
library(jsonlite)
library(xml2)
library(ggplot2)
theme_set(theme_bw())
library(tibble)
library(tidyr)
library(magrittr)
library(glue)
library(purrr)
library(rsnps)
library(data.table)
library(janitor)
library(stringr)
library(rsnps)The squared correlation between genetic markers is one way to estimate linkage disequilibrium (LD). LD has to be computed all the time - either for as an input for statistical methods or to summarise results.
However, accessing LD estimations quickly, for a specific population and in an automated way (e.g. with R) is suprisingly difficult.
In this blog post I am exploring how to do this efficiently.
Goal
At the end of this blog post, we want to know the genetic correlation between two or more markers in a specific human population, so that we can populate the locuszoom plot from the previous blog post with coloured dots.
For simplicity, I will use the terms correlation, squared correlation, r, r2 and LD interchangeably.
Two approaches
In principle, there are two ways of doing accessing LD:
- Download (or access) the genetic data from which you want to estimate your correlations + calculate the correlations using some efficient approach.
- Access precomputed LD estimations.
| Approach | Advantages | Downsides | Useful when… |
|---|---|---|---|
| (1) Local computation of LD | LD matrix can be quickly updated to new reference panels | Requires large computation and storage space (e.g. 1000 Genomes is >100 few GB large). | i) LD for a large set of SNPs is needed ii) LD from non-standard reference panel is needed. |
| (2) Access precomputed LD | not need for large computation and storage space. | limited to certain small sets of markers, limited to possibly outdated reference panels. | LD for a small set of SNPs is needed |
For now, I will focus on approach (2), and then explore approach (1) in a future blog post.
Spoiler: Using approach (2) does not get you far. It took me quite a while to gather all the solutions that are listed below, and yet there is not one perfect/ideal solution.
Our toy data
We will recycle the data from the previous blog post, where the focus was on extracting annotation using the package biomaRt. In this blog post, we will complete that locuszoom plot by adding the LD information.
## Data Source URL
url <- "https://portals.broadinstitute.org/collaboration/giant/images/2/21/BMI_All_ancestry.fmt.gzip"
## Import BMI summary statistics dat.bmi <- read_tsv(file = url) ##
## taking too long, let's use fread instead.
dat.bmi <- data.table::fread(url, verbose = FALSE)
## Rename some columns
dat.bmi <- dat.bmi %>% rename(SNP = SNPNAME, P = Pvalue)
## Extract region
dat.bmi.sel <- dat.bmi %>% slice(which.min(P))
dat.bmi.sel
## range region
range <- 5e+05
sel.chr <- dat.bmi.sel$CHR
sel.pos <- dat.bmi.sel$POS
data <- dat.bmi %>%
filter(CHR == sel.chr, between(POS, sel.pos -
range, sel.pos + range))
head(data)
(snp <- dat.bmi.sel$SNP)What we are interested in is the LD between our top SNP rs1421085 and all other 82 SNPs nearby.
This dataset has positions on build GRCh37, while most databases are on build GRCh38 by now.
sm <- rsnps::ncbi_snp_summary(snp) %>% separate(chrpos, c("chr", "pos"))
sel.pos == as.numeric(sm$pos)## [1] FALSELet’s quickly repeat what our primary goal is:
Extract the correlation between SNPs
- without downloading any data,
- fairly quick and
- in R.
1A. A solution that works: ldlink from NIH
ldlink is a website provided by NIH to easily (and programmatically) request LD estimates in population groups.
LD is estimated from Phase 3 of the 1000 Genomes Project and super- and subpopulations can be selected.
There are different ways to access the LD estimations (e.g. LDpair, LDmatrix, LDproxy) and the same modules are also available through the API.
To access the API, you need to register for a token (takes a few seconds).
MYTOKEN <- "a_mix_of_numbers_and_characters"Let’s look at two modules:
LDproxy
To get the LD between a SNP and its region.
First, access the API:
LDproxy_raw <- system(
glue::glue("curl -k -X GET 'https://ldlink.nci.nih.gov/LDlinkRest/ldproxy?var={snp}&pop=EUR&r2_d=r2&token={MYTOKEN}'"),
intern = TRUE
)Then, do a bit of data wrangling to get a tidy data frame:
LDproxy <- LDproxy_raw %>%
purrr::map(., function(x) stringr::str_split(x, "\t") %>%
unlist()) %>% ## remove all the tabs
do.call(rbind, .) %>% ## turn into a matrix
data.frame() %>% ## turn into a data frame
janitor::row_to_names(1) %>% ## make the first row the column names
rename(SNP = RS_Number) %>% ## rename RS_Number as SNP
mutate_at(vars(MAF:R2), function(x) as.numeric(as.character(x))) %>% ## turn MAF:R2 columns numeric
mutate(SNP = as.character(SNP)) ## turn SNP from a factor into a character
head(LDproxy)## SNP Coord Alleles MAF Distance Dprime R2
## 1 rs1421085 chr16:53800954 (T/C) 0.4324 0 1.0000 1.0000
## 2 rs11642015 chr16:53802494 (C/T) 0.4324 1540 1.0000 1.0000
## 3 rs62048402 chr16:53803223 (G/A) 0.4324 2269 1.0000 1.0000
## 4 rs1558902 chr16:53803574 (T/A) 0.4324 2620 1.0000 1.0000
## 5 rs55872725 chr16:53809123 (C/T) 0.4324 8169 1.0000 1.0000
## 6 rs56094641 chr16:53806453 (A/G) 0.4344 5499 0.9959 0.9839
## Correlated_Alleles RegulomeDB Function
## 1 T=T,C=C 5 NA
## 2 T=C,C=T 4 NA
## 3 T=G,C=A 5 NA
## 4 T=T,C=A 7 NA
## 5 T=C,C=T 4 NA
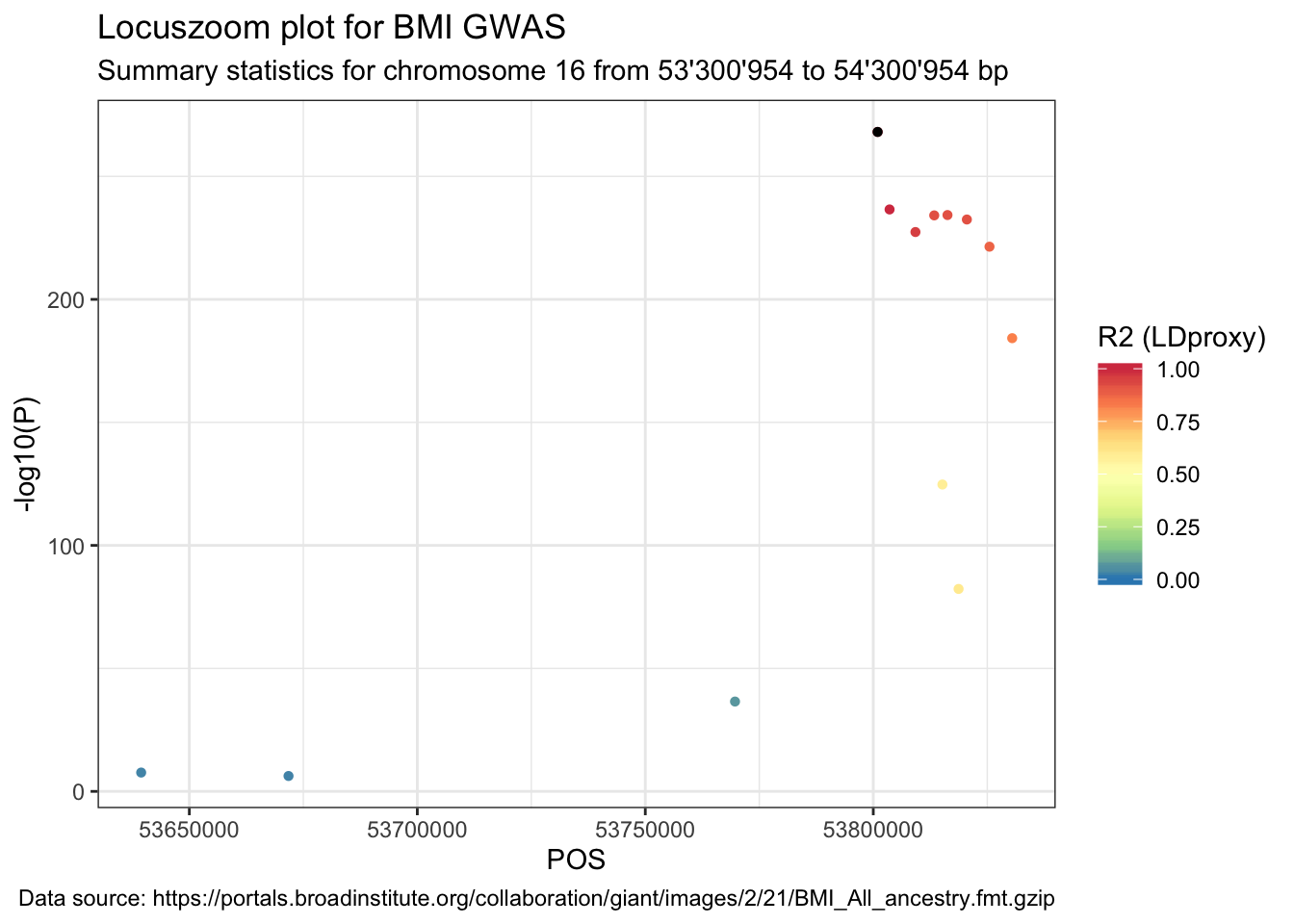
## 6 T=A,C=G 6 NANext, join the original summary stats data with the LDproxy data frame.
data_ldproxy <- data %>%
right_join(LDproxy, by = c("SNP" = "SNP"))Lastly, plot the summary statistics with the point colour indicating the R2.
ggplot(data = data_ldproxy) +
geom_point(aes(POS, -log10(P), color = R2), shape = 16) +
labs(
title = "Locuszoom plot for BMI GWAS",
subtitle = paste(
"Summary statistics for chromosome", sel.chr, "from",
format((sel.pos - range), big.mark = "'"), "to",
format((sel.pos + range), big.mark = "'"), "bp"
),
caption = paste("Data source:", url)
) +
geom_point(
data = data_ldproxy %>% filter(SNP == snp),
aes(POS, -log10(P)), color = "black", shape = 16
) +
scale_color_distiller("R2 (LDproxy)",
type = "div", palette = "Spectral",
limits = c(0, 1)
)
LDmatrix
LDmatrix module accesses the pairwise LD between a set of SNPs.
Again, first access the API:
snplist <- data %>%
filter(str_detect(SNP, "rs")) %>%
pull(SNP) %>%
paste(collapse = "%0A")
LDmatrix_raw <- system(
glue::glue("curl -k -X GET 'https://ldlink.nci.nih.gov/LDlinkRest/ldmatrix?snps={snplist}&pop=CEU%2BTSI%2BFIN%2BGBR%2BIBS&r2_d=r2&token={MYTOKEN}'"),
intern = TRUE
)- If you want to access the dprime (d’) values, write
r2_d=d. - If you want to access certain sub populations, let’s say CEU, TSI and FIN, concatenate them with
%Bin between:CEU%2BTSI%2BFIN.
Then, do a little data tidying:
LDmatrix <- LDmatrix_raw %>%
purrr::map(., function(x) stringr::str_split(x, "\t") %>%
unlist()) %>%
do.call(rbind, .) %>% ## turn into a matrix
data.frame() %>% ## turn into a data.frame
janitor::row_to_names(1) ## make the first line the column names
LDmatrix_long <- LDmatrix %>%
gather("SNP2", "R2", -RS_number) %>% ## from wide to long
rename(SNP = RS_number) %>% ## rename RS_number
mutate(R2 = as.numeric(R2)) %>% ## make R2 numeric
mutate_if(is.factor, as.character) ## make all factor columns characters## Warning: attributes are not identical across measure variables;
## they will be dropped## Warning: NAs introduced by coercionhead(LDmatrix_long)## SNP SNP2 R2
## 1 rs61754093 rs61754093 1.000
## 2 rs181111349 rs61754093 NA
## 3 rs199662749 rs61754093 NA
## 4 rs189080082 rs61754093 0.000
## 5 rs6499548 rs61754093 0.023
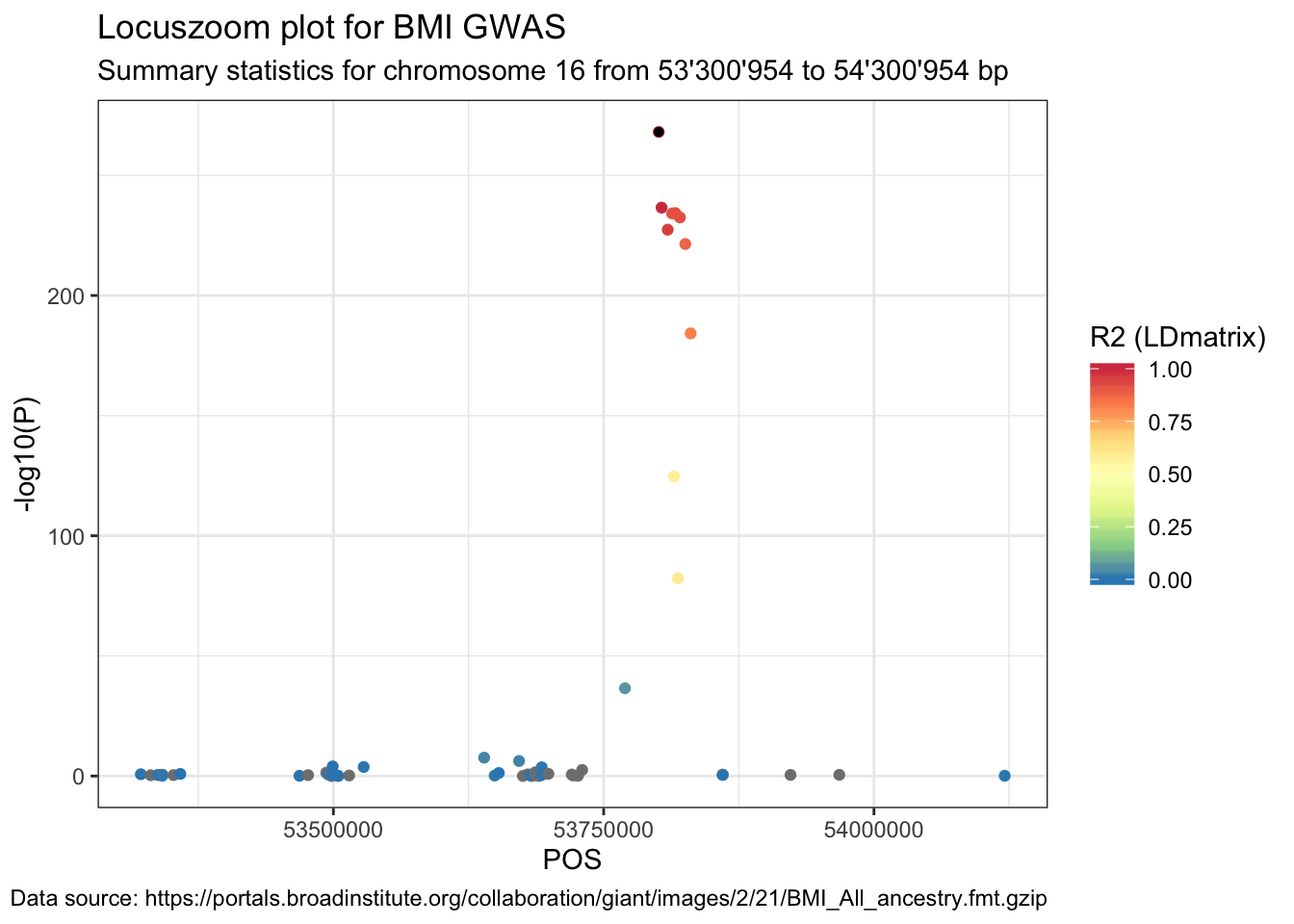
## 6 rs139704369 rs61754093 NANext, join the original summary stats data with the LDmatrix_long data frame.
data_ldmatrix <- data %>%
right_join(LDmatrix_long, by = c("SNP" = "SNP"))Lastly, plot the summary statistics with the point colour indicating the R2.
ggplot(data = data_ldmatrix %>% filter(SNP2 == snp)) +
geom_point(aes(POS, -log10(P), color = R2)) +
labs(
title = "Locuszoom plot for BMI GWAS",
subtitle = paste(
"Summary statistics for chromosome", sel.chr, "from",
format((sel.pos - range), big.mark = "'"), "to",
format((sel.pos + range), big.mark = "'"), "bp"
),
caption = paste("Data source:", url)
) +
geom_point(
data = data_ldmatrix %>% filter(SNP == snp & SNP2 == snp),
aes(POS, -log10(P)), color = "black", shape = 16
) +
scale_color_distiller("R2 (LDmatrix)",
type = "div", palette = "Spectral",
limits = c(0, 1)
)
1B. A solution that almost works: ensembl.org
The REST API of Ensembl can do a lot (see options here). For example access precomputed LD. The webpage even provides R code to do so, which is from where I copied some snippets below.
To access the rest API at ensembl, we need the following three packages loaded.
library(httr)
library(jsonlite)
library(xml2)What reference panels/population can we choose from?
Currently, the largest and hence most popular public reference panel is 1000 Genomes reference panel (1KG). The 26 populations of roughly 100 individuals each can be grouped into five super populations: African (AFR), American (AMR), European (EUR), South Asian (SAS), East Asian (EAS).
We can ask the ENSEMBL API from what populations reference panels are available. This will return us a data frame.
server <- "https://rest.ensembl.org"
ext <- "/info/variation/populations/homo_sapiens?filter=LD"
r <- GET(paste(server, ext, sep = ""), content_type("application/json"))
stop_for_status(r)
head(fromJSON(toJSON(content(r))))## description size
## 1 African Caribbean in Barbados 96
## 2 African Ancestry in Southwest US 61
## 3 Bengali in Bangladesh 86
## 4 Chinese Dai in Xishuangbanna, China 93
## 5 Utah residents with Northern and Western European ancestry 99
## 6 Han Chinese in Bejing, China 103
## name
## 1 1000GENOMES:phase_3:ACB
## 2 1000GENOMES:phase_3:ASW
## 3 1000GENOMES:phase_3:BEB
## 4 1000GENOMES:phase_3:CDX
## 5 1000GENOMES:phase_3:CEU
## 6 1000GENOMES:phase_3:CHBname stands for the population identifier. size refers to the number of individuals in the reference panel. Note that these are all populations with around 100 individuals (the correlation estimation will have an error that scales with the sample size). There are also the five super population available (although not listed here), simply replace the last three characters in name by EUR, AFR, AMR, EAS, SAS.
(From this blog post.)
We want the LD information, so that we can add this info to the locuszoom plot. But how do we know which population to pick? One way is to read up what kind of individuals were present. In our case - mostly Europeans (EUR). But we could also build some pooled LD matrix of different populations.
Now that we know which reference panel we want to use, we can use the different rest APIs.
Access LD between a SNP and its region
This API is described here.
The default window size is 500 kb. There are also thresholds for r2 (e.g. if you want to filter all SNPs with an r2 > 0.8).
The only input required is the SNP rsid, marked with {snp}.
snp## [1] "rs1421085"server <- "https://rest.ensembl.org"
ext <- glue::glue("/ld/human/{snp}/1000GENOMES:phase_3:EUR?")
## Window size in kb. The maximum allowed value for the window size is 500 kb. LD is computed for the given variant and all variants that are located within the specified window.
r <- GET(paste(server, ext, sep = ""), content_type("application/json"))
stop_for_status(r)
LD.SNP.region <- as_tibble(fromJSON(toJSON(content(r)))) %>%
unnest() %>%
mutate(r2 = as.numeric(r2))## Warning: `cols` is now required.
## Please use `cols = c(variation1, d_prime, r2, population_name, variation2)`head(LD.SNP.region)## # A tibble: 6 x 5
## variation1 d_prime r2 population_name variation2
## <chr> <chr> <dbl> <chr> <chr>
## 1 rs1421085 0.327013 0.0834 1000GENOMES:phase_3:EUR rs8043738
## 2 rs1421085 0.992084 0.409 1000GENOMES:phase_3:EUR rs2042031
## 3 rs1421085 0.846280 0.647 1000GENOMES:phase_3:EUR rs11642841
## 4 rs1421085 1.000000 0.957 1000GENOMES:phase_3:EUR rs9940128
## 5 rs1421085 0.999948 0.0552 1000GENOMES:phase_3:EUR rs73612011
## 6 rs1421085 0.907868 0.648 1000GENOMES:phase_3:EUR rs8057044As a result, LD.snp.region contains the r2 of our top SNP with all SNPs that were +/- 500 kb away.
What if we want the correlation between all SNPs?
Access LD matrix
For this, we need the rest API here.
We can calculate the LD matrix of a full region, max 1 Mb wide. For fast computation, we limit it to +/- 50 kb.
## Query region. A maximum of 1Mb is allowed.
ext <- glue::glue("/ld/human/region/{sel.chr}:{sel.pos - range/20}..{sel.pos + range/20}/1000GENOMES:phase_3:EUR?")
r <- GET(paste(server, ext, sep = ""), content_type("application/json"))
stop_for_status(r)
LD.matrix.region <- as_tibble(fromJSON(toJSON(content(r)))) %>%
unnest() %>%
mutate(r2 = as.numeric(r2))## Warning: `cols` is now required.
## Please use `cols = c(population_name, r2, variation2, variation1, d_prime)`head(LD.matrix.region)## # A tibble: 6 x 5
## population_name r2 variation2 variation1 d_prime
## <chr> <dbl> <chr> <chr> <chr>
## 1 1000GENOMES:phase_3:EUR 0.761 rs9933509 rs8057044 0.999998
## 2 1000GENOMES:phase_3:EUR 1 rs150763868 rs72803680 1.000000
## 3 1000GENOMES:phase_3:EUR 0.106 rs7206122 rs62033401 0.999940
## 4 1000GENOMES:phase_3:EUR 0.731 rs9935401 rs8057044 0.999997
## 5 1000GENOMES:phase_3:EUR 0.106 rs141816793 rs62033399 0.999943
## 6 1000GENOMES:phase_3:EUR 0.996 rs9941349 rs9931900 1.000000Access LD between a SNP and many other SNPs
The third and last option is to pass on a set of SNP rs ids, and access the LD among these. Implemented in the ENSEMBL API is only the LD between two SNPs, so we will have to extend this to many SNPs.
extract_ld <- function(SNP.id2 = NULL, SNP.id1 = NULL, POP = NULL) {
ext <- glue::glue("/ld/human/pairwise/{SNP.id1}/{SNP.id2}/") ## filter POP further down
server <- "https://rest.ensembl.org"
r <- GET(paste(server, ext, sep = ""), content_type("application/json"))
stop_for_status(r)
out <- as_tibble(fromJSON(toJSON(content(r)))) %>%
unnest() %>%
filter(stringr::str_detect(population_name, POP))
return(out)
}
## see futher down why intersect here
other.snps <- intersect(LD.SNP.region$variation2, data$SNP)
## cacluate LD for all other.snps SNPs
LD.matrix.snps <- purrr::map_df(other.snps, extract_ld, snp, "EUR") %>%
mutate(r2 = as.numeric(r2)) %>%
bind_rows() %>%
unnest()## Warning: `cols` is now required.
## Please use `cols = c(variation2, d_prime, variation1, r2, population_name)`
## Warning: `cols` is now required.
## Please use `cols = c(variation2, d_prime, variation1, r2, population_name)`
## Warning: `cols` is now required.
## Please use `cols = c(variation2, d_prime, variation1, r2, population_name)`## Warning: `cols` is now required.
## Please use `cols = c(d_prime, variation1, variation2, r2, population_name)`## Warning: `cols` is now required.
## Please use `cols = c(variation2, variation1, d_prime, population_name, r2)`## Warning: `cols` is now required.
## Please use `cols = c(variation1, d_prime, variation2, population_name, r2)`## Warning: `cols` is now required.
## Please use `cols = c(r2, population_name, d_prime, variation1, variation2)`## Warning: `cols` is now required.
## Please use `cols = c(population_name, r2, variation1, d_prime, variation2)`## Warning: `cols` is now required.
## Please use `cols = c(variation2, d_prime, variation1, r2, population_name)`## Warning: `cols` is now required.
## Please use `cols = c(d_prime, variation1, variation2, r2, population_name)`## Warning: `cols` is now required.
## Please use `cols = c()`LD.matrix.snps## # A tibble: 0 x 5
## # … with 5 variables: variation2 <chr>, d_prime <chr>, variation1 <chr>,
## # r2 <dbl>, population_name <chr>Calculate the LD matrix (LD.matrix.region) or the LD between SNP pairs (LD.matrix.snps) takes a lot of time!
Coloured locuszoom plot
For the locuszoom plot we need only the correlation between the top SNP and all other SNPs. So we join the object LD.SNP.region to data.
data_ensembl <- data %>%
full_join(LD.SNP.region, by = c("SNP" = "variation2"))ggplot(data = data_ensembl) +
geom_point(aes(POS, -log10(P), color = r2), shape = 16) +
labs(
title = "Locuszoom plot for BMI GWAS",
subtitle = paste("Summary statistics for chromosome", sel.chr, "from", format((sel.pos - range), big.mark = "'"), "to", format((sel.pos + range), big.mark = "'"), "bp"),
caption = paste("Data source:", url)
) +
geom_point(
data = data_ensembl %>% filter(SNP == "rs1421085"),
aes(POS, -log10(P)), color = "black", shape = 16
) +
scale_color_distiller("R2 (ensembl)", type = "div", palette = "Spectral", limits = c(0, 1))## Warning: Removed 205 rows containing missing values (geom_point).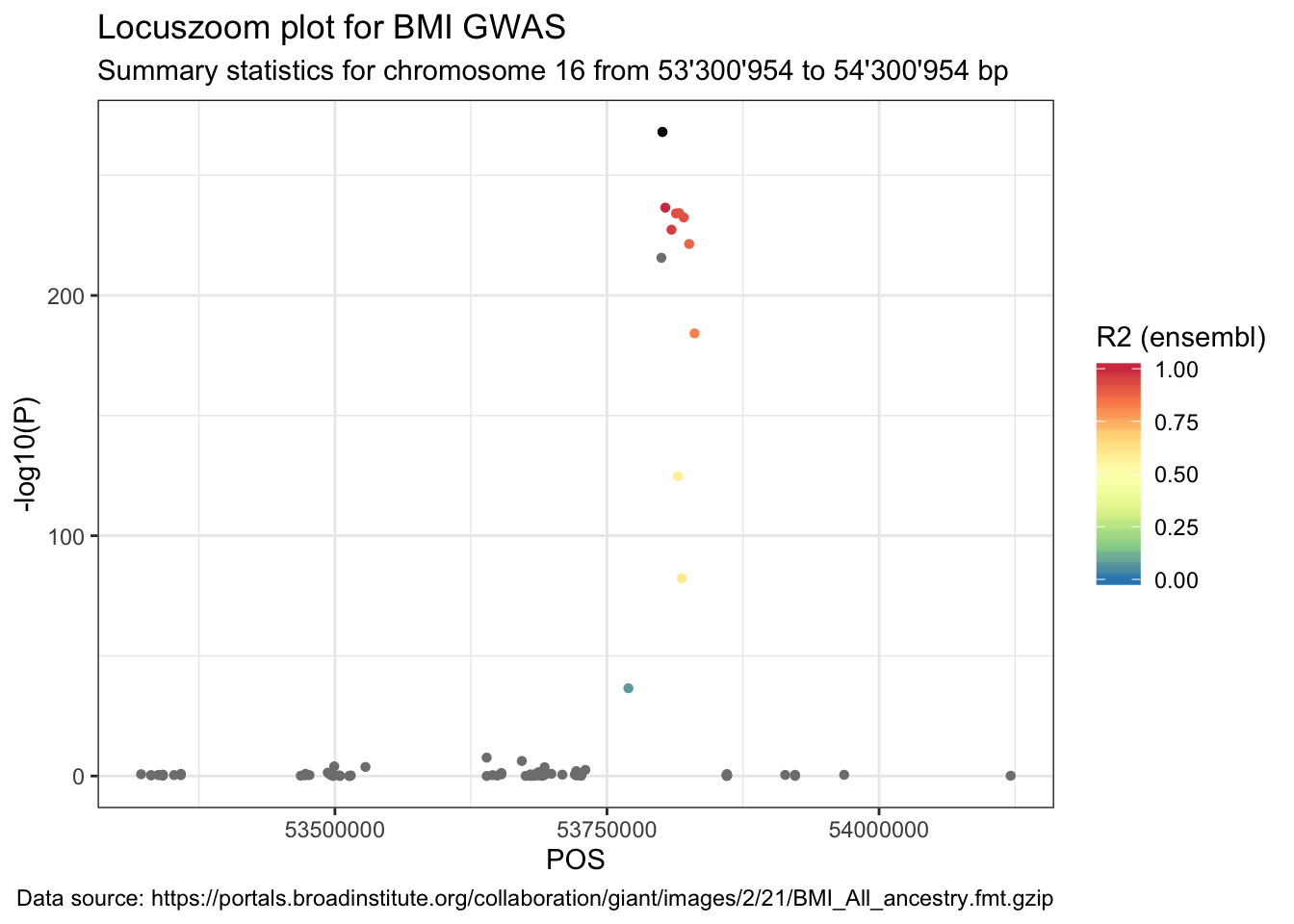
2. Solutions that work half-through
SNPsnap
- SNPsnap: https://data.broadinstitute.org/mpg/snpsnap/database_download.html
- uses a limited set of 1KG populations (EUR, EAS, WAFR).
API provided by sph.umich
- API
- uses limited set of 1KG populations (ALL, EUR)
- see github issue
3. A solution that does not work
rsnps::ld_search
A perfect solution would have been the function ld_search from R package rsnps. It has arguments to choose the reference panel, the population, the distance from the focal SNP.
The problem is that it only uses old reference panels (HapMap and 1KG-phase1). Meaning, many newer reference panel populations are left out.
But the main problem is, that the broad institute has taken down the snap server that ld_search used to access (see github issue); hence ld_search is defunct.
Conclusion
The
ldlink API with the LDproxy module seems the most perfect solution for now.
This will probably change with changing technology and larger reference panels.
Session Info
sessionInfo()## R version 3.5.1 (2018-07-02)
## Platform: x86_64-apple-darwin15.6.0 (64-bit)
## Running under: macOS 10.14.5
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] janitor_1.2.0 data.table_1.12.2 rsnps_0.3.2.9121
## [4] glue_1.3.1.9000 magrittr_1.5 xml2_1.2.0
## [7] jsonlite_1.6 httr_1.4.0 forcats_0.4.0.9000
## [10] stringr_1.4.0 dplyr_0.8.3.9000 purrr_0.3.2
## [13] readr_1.3.1 tidyr_0.8.3.9000 tibble_2.1.3
## [16] ggplot2_3.2.0.9000 tidyverse_1.2.1
##
## loaded via a namespace (and not attached):
## [1] tidyselect_0.2.5 xfun_0.8 haven_2.1.1 lattice_0.20-38
## [5] colorspace_1.4-1 vctrs_0.2.0.9000 generics_0.0.2 htmltools_0.3.6
## [9] yaml_2.2.0 utf8_1.1.4 XML_3.98-1.20 rlang_0.4.0
## [13] pillar_1.4.2 httpcode_0.2.0 withr_2.1.2 modelr_0.1.4
## [17] readxl_1.3.1 plyr_1.8.4 munsell_0.5.0 blogdown_0.13
## [21] gtable_0.3.0 cellranger_1.1.0 rvest_0.3.4 evaluate_0.14
## [25] knitr_1.23 curl_3.3 fansi_0.4.0 triebeard_0.3.0
## [29] urltools_1.7.3 broom_0.5.2 Rcpp_1.0.1 scales_1.0.0
## [33] backports_1.1.4 hms_0.4.2 digest_0.6.20 stringi_1.4.3
## [37] bookdown_0.11 grid_3.5.1 cli_1.1.0 tools_3.5.1
## [41] lazyeval_0.2.2 crul_0.8.0 crayon_1.3.4 pkgconfig_2.0.2
## [45] zeallot_0.1.0 ellipsis_0.2.0.1 lubridate_1.7.4 assertthat_0.2.1
## [49] rmarkdown_1.13 rstudioapi_0.10 R6_2.4.0 icon_0.1.0
## [53] nlme_3.1-140 compiler_3.5.1)